Tamara Simakova
2014-04-15 06:53:58 UTC
Hello!
I'm using BedTools on the main Galaxy server. I try to use multiinter
function to compare two bed files, but there is an error in the resulted
file. Some coordinates that are present in both files are marked as they
present only in one file. The bed-files format is correct. What could be
the problem?
Thanks,
Tamara Simakova
-------------- next part --------------
An HTML attachment was scrubbed...
URL: <http://lists.bx.psu.edu/pipermail/galaxy-user/attachments/20140415/5459e2b5/attachment-0001.html>
-------------- next part --------------
A non-text attachment was scrubbed...
Name: Bedtools_error.jpg
Type: image/jpeg
Size: 555554 bytes
Desc: not available
URL: <Loading Image...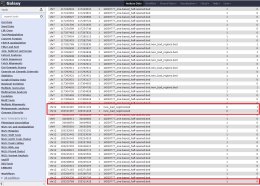 >
>
-------------- next part --------------
A non-text attachment was scrubbed...
Name: IAD39777_one-based_half-opened.bed
Type: application/octet-stream
Size: 1357 bytes
Desc: not available
URL: <http://lists.bx.psu.edu/pipermail/galaxy-user/attachments/20140415/5459e2b5/attachment-0002.obj>
-------------- next part --------------
A non-text attachment was scrubbed...
Name: new_bad_regions.bed
Type: application/octet-stream
Size: 442 bytes
Desc: not available
URL: <http://lists.bx.psu.edu/pipermail/galaxy-user/attachments/20140415/5459e2b5/attachment-0003.obj>
I'm using BedTools on the main Galaxy server. I try to use multiinter
function to compare two bed files, but there is an error in the resulted
file. Some coordinates that are present in both files are marked as they
present only in one file. The bed-files format is correct. What could be
the problem?
Thanks,
Tamara Simakova
-------------- next part --------------
An HTML attachment was scrubbed...
URL: <http://lists.bx.psu.edu/pipermail/galaxy-user/attachments/20140415/5459e2b5/attachment-0001.html>
-------------- next part --------------
A non-text attachment was scrubbed...
Name: Bedtools_error.jpg
Type: image/jpeg
Size: 555554 bytes
Desc: not available
URL: <Loading Image...
 >
>-------------- next part --------------
A non-text attachment was scrubbed...
Name: IAD39777_one-based_half-opened.bed
Type: application/octet-stream
Size: 1357 bytes
Desc: not available
URL: <http://lists.bx.psu.edu/pipermail/galaxy-user/attachments/20140415/5459e2b5/attachment-0002.obj>
-------------- next part --------------
A non-text attachment was scrubbed...
Name: new_bad_regions.bed
Type: application/octet-stream
Size: 442 bytes
Desc: not available
URL: <http://lists.bx.psu.edu/pipermail/galaxy-user/attachments/20140415/5459e2b5/attachment-0003.obj>